Patología suprarrenal
Autores
Alba Martín González. Endocrinología y Nutrición. | Javier Tejada Montes. Medicina Interna.
Asesora
María Calatayud Gutiérrez. Endocrinología y Nutrición.
1. Síndrome de Cushing
1.1. Definición y etiología
El síndrome de Cushing (SC) engloba todo el conjunto de signos y síntomas derivados de la exposición prolongada a niveles inapropiadamente elevados de glucocorticoides (GC) (tabla 1).
1.2. Clínica
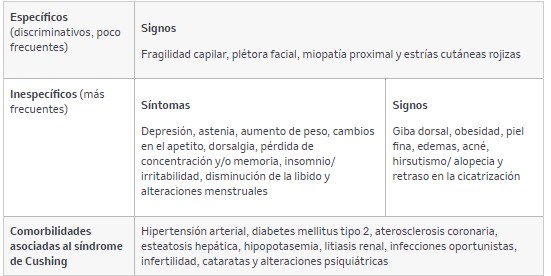
Las manifestaciones clínicas y comorbilidades asociadas al síndrome de Cushing se presentan en la tabla 2.
Tabla 1. Principales causas del síndrome de Cushing

Tabla 2. Manifestaciones clínicas y comorbilidades asociadas al síndrome de Cushing
1.3. Diagnóstico
1.3.1. Población a estudio
El diagnóstico certero de Cushing es un reto debido a las importantes interferencias en los diferentes test utilizados. El estudio está indicado en:
- Pacientes con varios y progresivos signos y síntomas sospechosos de SC (tabla 2), especialmente los más específicos.
- Pacientes con comorbilidades asociadas inusuales para su edad o de difícil control (diabetes mellitus, hipertensión arterial y osteoporosis en pacientes jóvenes).
- Todos los pacientes con incidentalomas suprarrenales.
1.3.2. Consideraciones previas a la realización del test de cribado
- Descartar el posible uso exógeno de corticoides (incluidos tópicos y/o inhalados, acetato de megestrol u otras progestinas con alguna actividad GC intrínseca).
- Se desaconseja estudiar a pacientes durante la fase de inestabilidad/estrés psicológico o físico importante o enfermedad aguda.
- Descartar causas de pseudocushing (tabla 3).
1.3.3. Pruebas diagnósticas
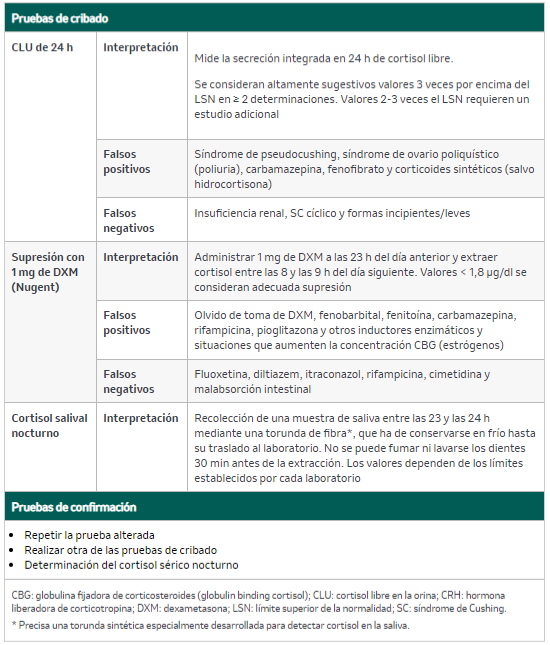
Se recomienda un estudio escalonado (figura 1). La confirmación bioquímica exige al menos dos pruebas alteradas extraídas en pacientes con sospecha clínica tras descartar interferencias. Los pacientes con alta sospecha clínica y resultados en los test diagnósticos iniciales compatibles con SC deben ser valorados por un especialista en Endocrinología (tabla 4).
1.4. Tratamiento
El tratamiento del SC, en caso de localizarse el origen y de que no exista contraindicación, es siempre quirúrgico. El tratamiento médico solo se emplea antes de cirugía o en casos en los que está contraindicada debiendo ser indicado y seleccionado por un facultativo especializado en SC.
Tabla 3. Causas frecuentes de pseudocushing
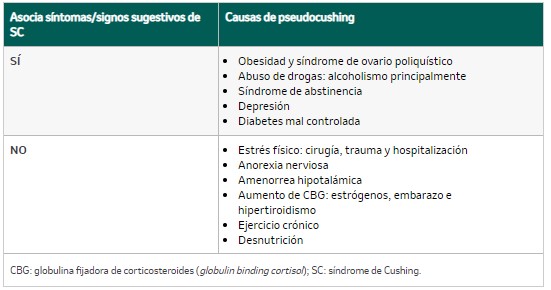
Tabla 4. Pruebas bioquímicas en el estudio del síndrome de Cushing
2. Insuficiencia suprarrenal
2.1. Definición y etiología
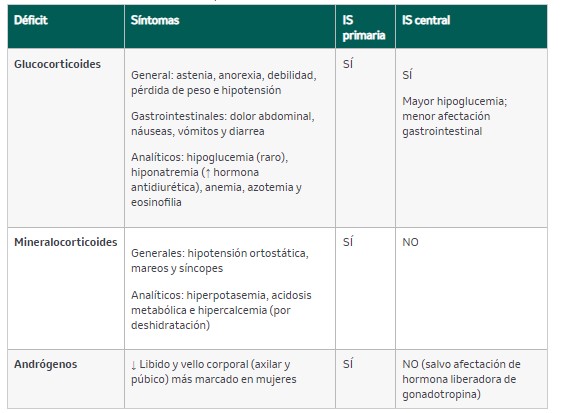
La insuficiencia suprarrenal (IS) es un trastorno que se caracteriza por déficit de GC (tabla 5). Según el origen del déficit, puede clasificarse en IS primaria, debida a procesos que afectan a la secreción de gluco- y mineralocorticoides en la corteza adrenal, e IS central, que se debe a procesos que afectan a la secreción de corticotropina a nivel hipofisario (IS secundaria) o a trastornos hipotalámicos que afectan a la secreción de la hormona liberadora de corticotropina (CRH) (IS terciaria).
2.2. Clínica
Generalmente se instaura de manera lenta y suele aparecer coincidiendo consituaciones de estrés físico o enfermedades intercurrentes. Algunos síntomas dependen del origen del déficit; así, los síntomas derivados del déficit de mineralocorticoides y de la hiperpigmentación debida al aumento de ACTH solo ocurren en las formas de IS primaria. Las principales manifestaciones clínicas se recogen en la tabla 6.
Figura 1. Algoritmo diagnóstico del síndrome de Cushing.
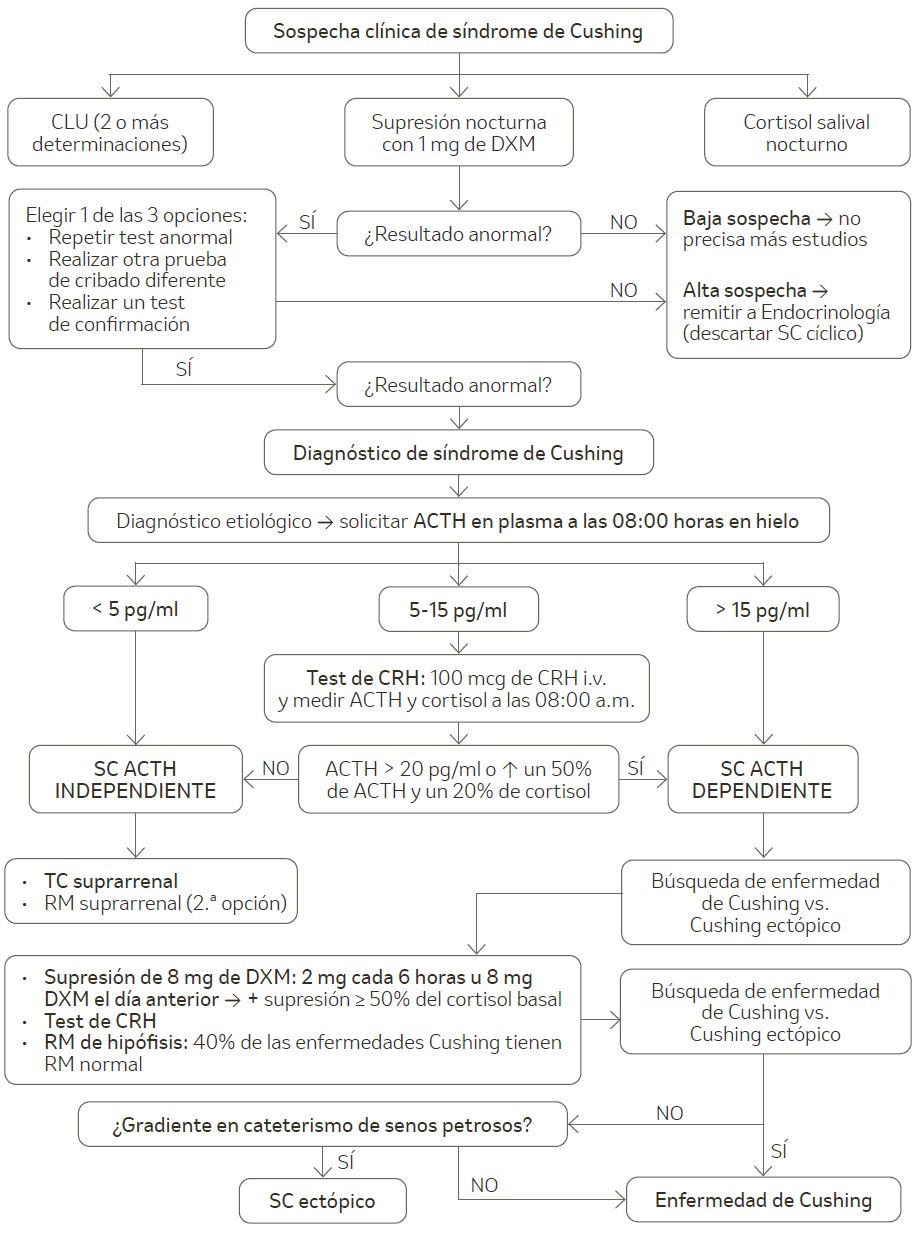
ACTH: hormona adrenocorticotropa; CLU: cortisol libre en la orina; DXM: dexametasona; CRH: hormona liberadora de corticotropina; RM: resonancia magnética; SC: síndrome de Cushing; TC: tomografía computarizada.
Tabla 5. Etiología de la insuficiencia suprarrenal

Tabla 6. Clínica asociada a insuficiencia suprarrenal
2.3. Diagnóstico
La evaluación diagnóstica en un paciente con sospecha o riesgo de IS incluye:
- Confirmar la existencia de una IS y nivel de eje afectado (figura 2).
- Identificar la causa responsable (tabla 5). Una vez confirmada la IS, se debe buscar el origen (primaria frente a central) mediante la determinación de la ACTH basal (figura 3).
2.4. Tratamiento de la insuficiencia suprarrenal crónica
2.4.1. Tratamiento sustitutivo con glucocorticoides
-
Hidrocortisona (Hidroaltesona® 20 mg): terapia de elección en el adulto. Se recomiendan dosis totales de 15-25 mg/día o 12 mg/m2 divididas en dos o tres tomas diarias para simular el ritmo circadiano del cortisol:
- Dos dosis: 2/3 de la dosis total diaria al levantarse y 1/4 en la merienda.
- Tres dosis: 1/2 de la dosis total diaria al levantarse, 1/4 al mediodía y 1/4 en la merienda. En la IS central se recomiendan dosis inferiores (10-15 mg/día o 6-8 mg/m2 divididas en 2-3 tomas/día).
Figura 2. Diagnóstico de insuficiencia suprarrenal.
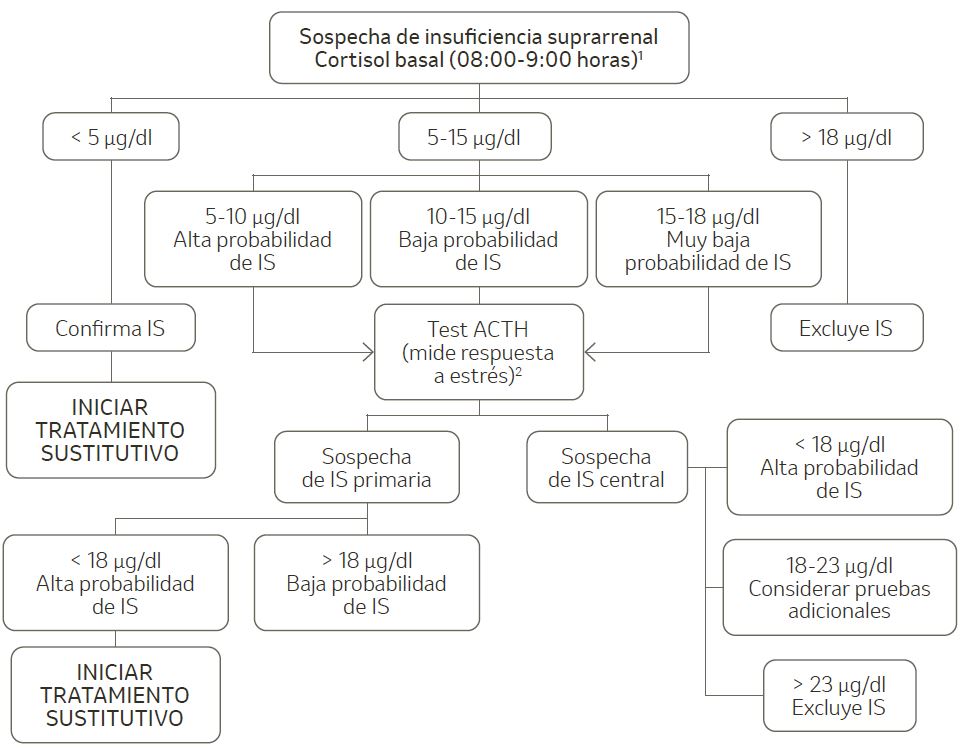
ACTH: hormona adrenocorticotropa; IS: insuficiencia suprarrenal.
Fuente: Adaptado de De Miguel. Endocrinol Nutr. 2014.
1 Se debe retirar cualquier corticoide exógeno (incluidos los tópicos o inhalados).
2 El test de estímulo con ACTH no es una prueba confirmatoria del diagnóstico de IS, sino una evaluación de la respuesta al estrés mediante la respuesta de la corteza adrenal a la administración de ACTH. Tiene poca sensibilidad en la IS parcial o en IS de reciente instauración (inferior a 4-6 semanas en cirugía hipofisaria o > 9- 12 meses en radioterapia); en estas situaciones, pueden valorarse otros test más específicos.
Figura 3. Etiología de la insuficiencia suprarrenal.
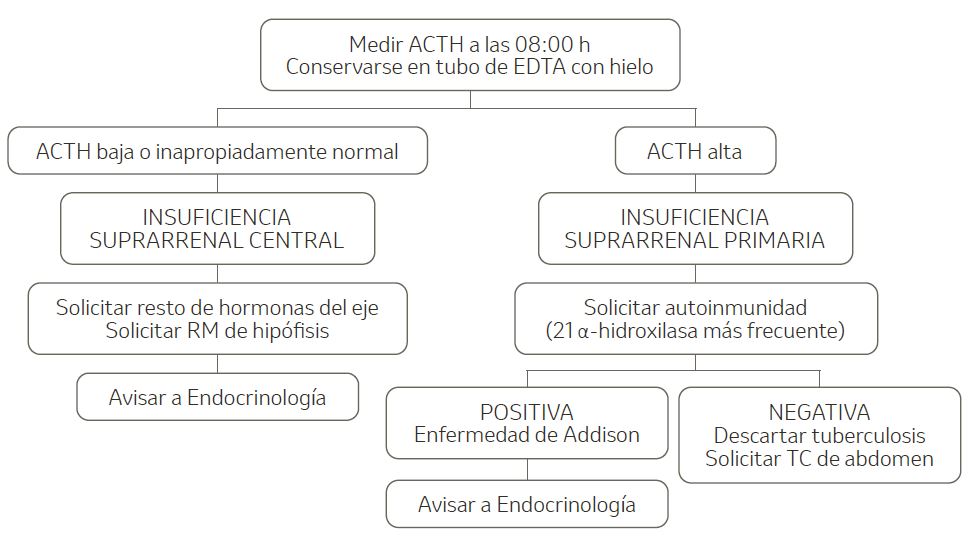
ACTH: hormona adrenocorticotropa; EDTA: ácido etilenodiaminatetraacético; RM: resonancia magnética; TC: tomografía computarizada.
Como alternativa pueden utilizarse:
- Dexametasona: dosis de 0,25-0,75 mg en una sola toma, preferiblemente en el desayuno; en la IS secundaria, dosis de 0,25-0,5 mg (dosis medias de 0,25 mg).
- Prednisona: dosis de 2,5-7,5 mg (dosis medias de 5 mg) en una sola toma, por la mañana; en la IS secundaria, dosis de 2,5-5 mg (dosis medias de 2,5 mg) en una sola toma.
2.4.2. Tratamiento sustitutivo con mineralocorticoides
Está indicado únicamente en la IS primaria y no siempre es necesario (varía en función del paciente y de la evolución): fludrocortisona (Astonin® 0,1 mg) a dosis de 0,05- 0,2 mg (dosis diaria habitual de 0,1 mg) en una sola toma al levantarse. El desarrollo de hipertensión, la aparición de edemas y la hipopotasemia es signo de reemplazo excesivo de mineralocorticoides, lo que obliga a un ajuste de dosis.
2.5. Manejo del paciente con insuficiencia suprarrenal en situación de estrés o terapia corticoide crónica
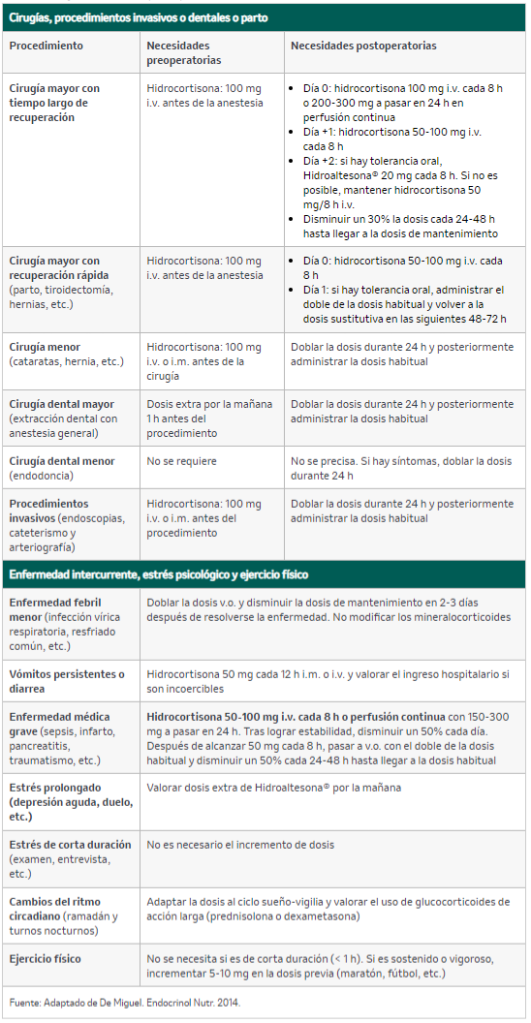
Existe una gran variación en la producción de cortisol en pacientes sanos sometidos a estrés, por lo que resulta difícil predecir de forma exacta las necesidades de los pacientes con IS ante estas situaciones y prevenir el desarrollo de una crisis suprarrenal. De forma generalizada, se debe actuar según la tabla 7.
Tabla 7. Manejo de corticoterapia en paciente en situación de estrés
2.6. Crisis suprarrenal aguda o crisis addisoniana
Consiste en un desequilibrio entre la producción y las necesidades fisiológicas de cortisol exógeno. Es una situación de riesgo vital y suele producirse ante situaciones de estrés en pacientes con IS conocida o como forma de debut. Resulta esencial su identificación, así como su prevención ante situaciones precipitantes (como se especifica en la tabla 7).
2.6.1. Clínica y diagnóstico
Se debe sospechar ante un paciente con inestabilidad hemodinámica, signos de deshidratación, clínica constitucional, hipoglucemia, hiponatremia, hiperpotasemia y/o eosinofilia, dolor abdominal que asocia sensación nauseosa y/o vómitos; que tenga algún factor de riesgo para desarrollar una crisis suprarrenal o, en su defecto, presente datos de hiperpigmentación, principalmente en flexuras. Recientemente se han propuesto unos criterios para el diagnóstico (tabla 8).
2.6.2. Manejo de la crisis addisoniana
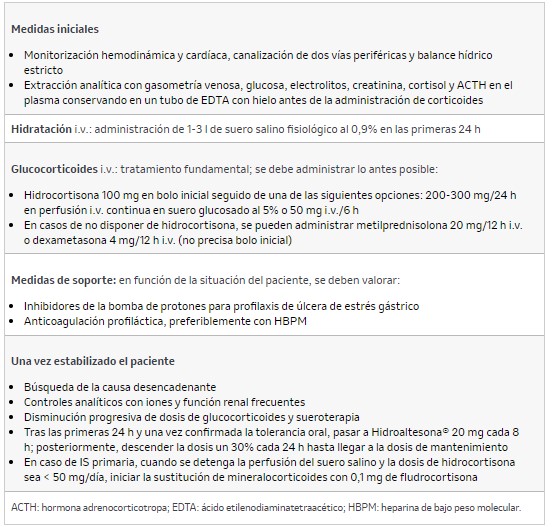
Ante un paciente con sospecha clínica de IS, hay que iniciar el tratamiento a pesar de no disponer de una confirmación analítica. La base es el tratamiento inmediato con hidrocortisona intravenosa, pero la sustitución de mineralocorticoides no es necesaria de forma aguda (el efecto retenedor de sodio de la fludrocortisona tarda varios días en manifestarse y las dosis altas de estrés de GC tienen efecto mineralocorticoide). El manejo general se recoge en la tabla 9.
2.6.3. Insuficiencia adrenal funcional, relativa o relacionada con la enfermedad crítica
Se refiere a la producción de cortisol en niveles subóptimos para las demandas totales del organismo en pacientes con enfermedad aguda grave. Su sintomatología es similar a la de cualquier IS, pero no hay consenso sobre los criterios de diagnóstico; los niveles de cortisol «óptimos» del paciente crítico no están estandarizados ni tampoco la dosis de ACTH que debe utilizarse en estos pacientes para comprobar una adecuada respuesta al estrés. Por otro lado, los diferentes estudios realizados indican que el uso de GC, preferiblemente la hidrocortisona, podría ayudar a una resolución más rápida del shock séptico, pero con un efecto mínimo o nulo sobre la mortalidad.
La Sociedad Europea de Medicina de Cuidados Intensivos (EISCM) recomienda el uso de corticoides en determinadas situaciones (ver cap. 3. Shock).
Tabla 8. Criterios diagnósticos de insuficiencia suprarrenal aguda*

Tabla 9. Manejo de la crisis addisoniana
2.7. Riesgo de supresión de eje y actitud para la suspensión del tratamiento crónico corticoide
Se debe valorar el riesgo de supresión de corticoides de cada individuo (tabla 10).
Para el descenso de la corticoterapia prolongada, es necesario:
- Valorar el riesgo de supresión del eje hipotálamo-hipófisis-adrenal (HHA) y de crisis adrenal.
- Proceder siempre que sea posible a un descenso gradual de la dosis de corticoide hasta lograr dosis fisiológicas (≤ 5 mg de prednisona o equivalentes).
- El tratamiento a días alternos puede promover la recuperación del eje en algunos pacientes.
- Durante el descenso progresivo de dosis, se debe vigilar la aparición de síntomas, que pueden deberse a:
- Hipocortisolismo: demostrado a través de alteraciones en el eje HHA mediante pruebas de laboratorio.
- Dependencia: suele asociar mejoría tras aumentar la dosis, aun en ausencia de alteraciones en el eje HHA en las pruebas de laboratorio.
- Reactivación de la enfermedad de base.
No hay un algoritmo ni una pauta exactos para el descenso de la corticoterapia que haya demostrado más eficacia y/o seguridad; en la tabla 11 se recoge un ejemplo de pauta de descenso. La equivalencia de los corticoides se puede ver en la tabla 12.
Tabla 10. Riesgo de supresión de corticoides

Tabla 11. Ejemplo de esquema de descenso de la corticoterapia en pacientes con riesgo de supresión de eje
3. Hiperaldosteronismo primario
3.1. Definición y etiología
Engloba el conjunto de trastornos intrínsecos a la glándula suprarrenal en los que la producción de aldosterona es inapropiadamente elevada y relativamente autónoma de los principales mecanismos reguladores de su secreción (angiotensina II y concentración de potasio plasmático). Las principales causas se recogen en la tabla 13.
Tabla 12. Equivalencia de corticoides
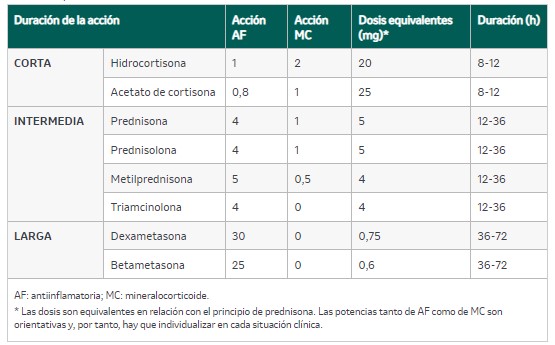
Tabla 13. Causas del hiperaldosteronismo primario

3.2. Clínica
- Hipertensión arterial: es el hallazgo clínico principal, y frecuentemente resulta refractaria. Constituye una causa infradiagnosticada de hipertensión arterial secundaria (en torno al 5-13% de los casos) y resulta imprescindible su reconocimiento por la mayor morbimortalidad cardiovascular asociada.
- Hipopotasemia: espontánea o inducida por diuréticos, tiene baja sensibilidad para el diagnóstico. En caso de adenomas suprarrenales, es más grave y suele darse en menores de 50 años.
- Otras alteraciones analíticas incluyen la alcalosis metabólica y, menos frecuentemente, hipernatremia e hipomagnesemia.
- Ausencia de edemas por fenómeno de «escape de la aldosterona».
- Comorbilidades asociadas: incremento en la tasa de morbimortalidad cardiovascular (hipertrofia del ventrículo izquierdo, infarto agudo de miocardio, insuficiencia cardíaca, fibrilación auricular e ictus), así como mayor riesgo de enfermedad renal crónica, síndrome de apneas-hipopneas del sueño, síndrome metabólico y diabetes mellitus.
3.3. Diagnóstico
3.3.1. Población a estudio
El despistaje se recomienda únicamente en poblaciones de riesgo (tabla 14).
3.3.1.1. Test diagnóstico
La determinación de la ratio aldosterona-renina (RAR) plasmática es la prueba indicada para el cribado del hiperaldosteronismo primario; sin embargo, no está exenta de falsos positivos y negativos y los puntos de corte son variables. Para su medición se deben tener en cuenta las consideraciones que aparecen en la tabla 15.
3.3.2. Diagnóstico de confirmación y determinación del subtipo
El algoritmo diagnóstico del hiperaldosteronismo primario se presenta en la figura 4.
3.4. Tratamiento
El tratamiento del hiperaldosteronismo primario se muestra en la tabla 16.
Tabla 14. Grupos de pacientes con alta prevalencia de hiperaldosteronismo

Tabla 15. Medición e interpretación de la ratio aldosterona-renina

Tabla 16. Tratamiento del hiperaldosteronismo primario

Figura 4. Algoritmo diagnóstico del hiperaldosteronismo primario.
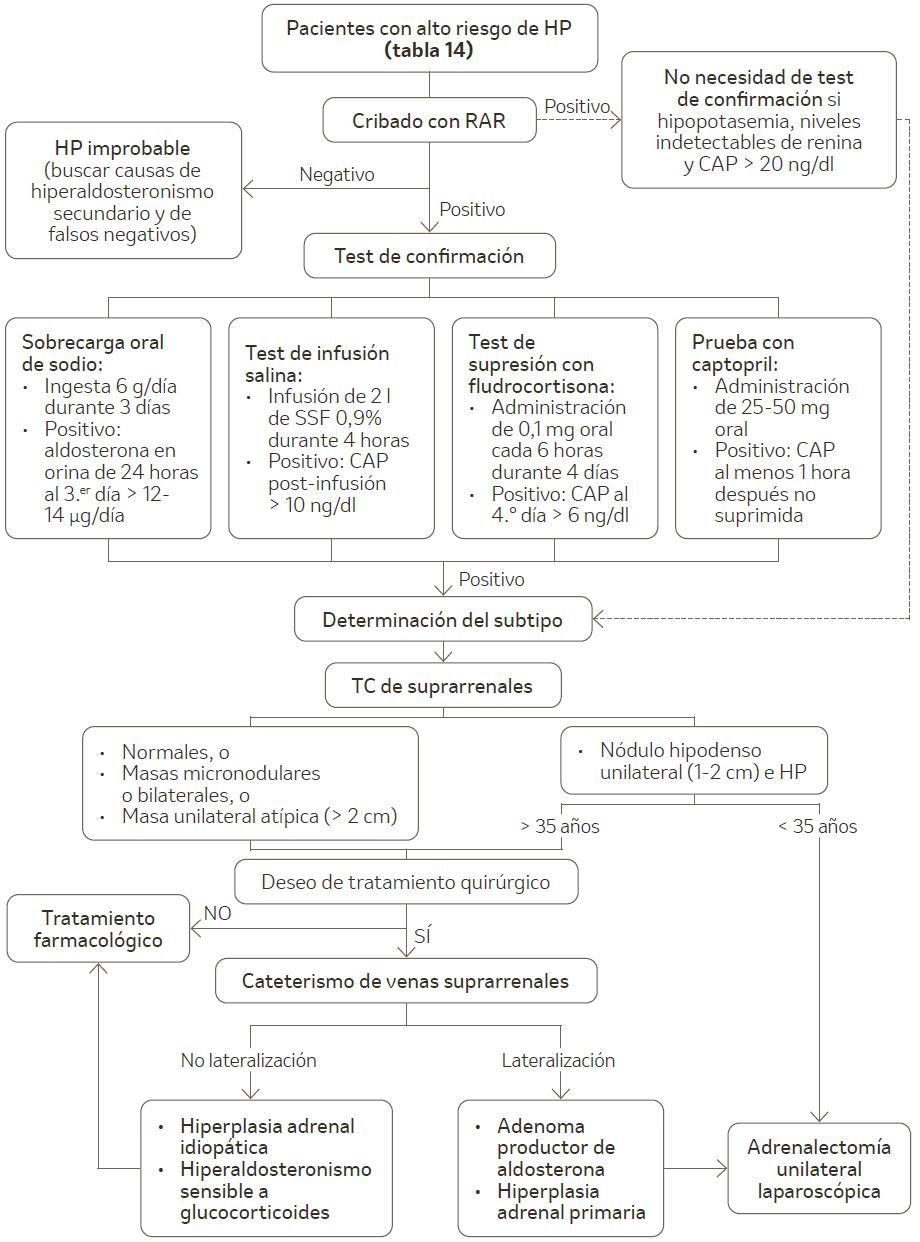
CAP: concentración de aldosterona plasmática; HP: hiperaldosteronismo primario; RAR: ratio aldosterona-renina; SSF: suero salino fisiológico; TC: tomografía computarizada.
4. Hipoaldosteronismo
Incluye los trastornos causados por déficit en la producción de aldosterona y/o resistencia tisular a ella. Como consecuencia, se producen hiperpotasemia (especialmente si asocia insuficiencia renal o uso de diuréticos ahorradores de potasio) y acidosis metabólica con anión GAP normal (denominada acidosis tubular renal tipo IV). Por el contrario, la hiponatremia es poco común debido a mecanismos compensadores, y su presencia debe hacer sospechar una IS primaria subyacente.
4.1. Hipoaldosteronismo hiporreninémico
Se caracteriza por la disminución en la liberación de renina y/o angiotensina II. Es más común en pacientes con nefropatía diabética debido a una lesión del aparato yuxtaglomerular; sin embargo, existen también otras causas, como nefropatías túbulo- intersticiales crónicas, nefroangiosclerosis hipertensiva, glomerulonefritis agudas, infección por el virus de la inmunodeficiencia humana (VIH), cirrosis hepática y fármacos (inhibidores de la calcineurina [ciclosporina], antiinflamatorios no esteroideos [AINE] e inhibidores de la enzima convertidora de angiotensina [IECA]/antagonistas del receptor de angiotensina II [ARA-II]).
La fludrocortisona puede ser efectiva en estos casos (dosis de 0,2-1 mg/día, más altas que en la IS primaria por un posible componente añadido de resistencia a la aldosterona); no obstante, puede empeorar la hipertensión arterial y/o los edemas. En estos pacientes una dieta pobre en potasio asociada al uso de un diurético tiazídico o de asa podrían ser suficientes para controlar la hiperpotasemia.
4.2. Hipoaldosteronismo hiperreninémico
Se produce como consecuencia de un defecto en la síntesis de aldosterona con un aumento compensatorio en la secreción de renina. Entre las principales causas se encuentran: IS primaria, fármacos (heparina sódica y de bajo peso molecular por efecto tóxico directo dosis-independiente sobre la capa glomerular), pacientes críticos (debido a expansión de volumen, hipersecreción de ACTH e hipercortisolemia) y enfermedades hereditarias (déficit de aldosterona-sintasa o de 21-hidroxilasa).
4.3. Pseudohipoaldosteronismo (resistencia a la aldosterona)
- Diuréticos ahorradores de potasio (espironolactona, eplerenona, amilorida y triamtereno) y ciertos antibióticos (trimetroprima o pentamidina) por inhibición del canal de sodio del túbulo colector (lugar donde actúa la aldosterona).
- Trastornos hereditarios (pseudohipoaldosteronismo tipo 1).
5. Feocromocitoma
5.1. Definición y clasificación
Es un tumor neuroendocrino poco frecuente productor de catecolaminas (adrenalina, noradrenalina y/o dopamina) que procede de las células cromafines de la médula adrenal. Constituye aproximadamente el 80-85% de los tumores originados del sistema nervioso simpático.
Las formas esporádicas son las más frecuentes (60-70%); en ocasiones también puede asociarse a trastornos hereditarios, entre los que se incluyen el síndrome de von Hippel-Lindau (10-20%), la neoplasia endocrina múltiple tipo 2 (50%) y la neurofibromatosis tipo 1 (2-3%).
5.2. Clínica
Está presente en aproximadamente el 50% de los casos, principalmente en forma de cuadros paroxísticos por la liberación episódica de catecolaminas (crisis adrenérgicas), que pueden ser espontáneos o precipitados por ciertos factores, entre los que se incluyen el ejercicio, algunos procedimientos (colonoscopia y angiografía), inducción de anestesia, ciertos alimentos que contienen tiramina o fármacos (β-bloqueantes, simpaticomiméticos, antidepresivos tricíclicos, GC, metoclopramida o inhibidores de monoaminooxidasa). Las principales manifestaciones clínicas se recogen en la tabla 17.
5.3. Diagnóstico
5.3.1. Población a estudio
Los grupos de pacientes con sospecha de feocromocitoma se muestran en la tabla 18.
5.3.1.1. Test diagnóstico
Se elige en función de la probabilidad pretest (figura 5). Los test empleados presentan valores predictivos negativos cercanos al 100%. Los fármacos/situaciones que pueden incrementar los niveles de catecolaminas y metanefrinas se pueden ver en la tabla 19.
Tabla 17. Tratamiento del hiperaldosteronismo primario

Tabla 18. Grupos de pacientes con sospecha de feocromocitoma

Figura 5. Algoritmo diagnóstico del feocromocitoma.
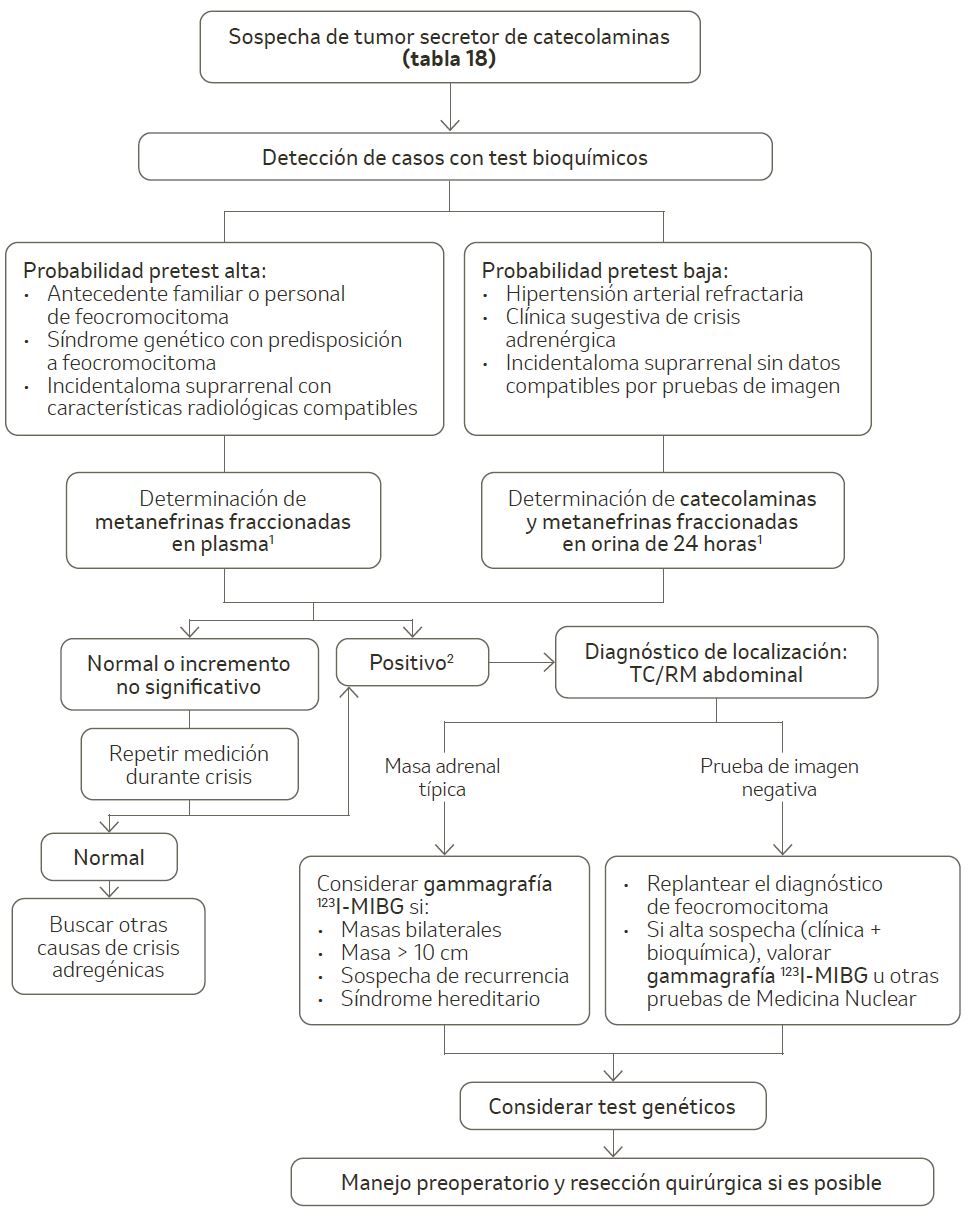
123I-MIBG: 123I-metayodobencilguanidina; RM: resonancia magnética; TC: tomografía computarizada.
1 Condiciones para obtención de muestra:
- Suspensión de fármacos (tabla 19) al menos 2 semanas antes
- Extracción de sangre en ayunas y después de 20-30 minutos de reposo en decúbito supino
- Dieta libre de aminas los 3-5 días previos a obtención de orina
2 Se considera un test positivo si se objetivan incrementos de cualquier metabolito dos veces por encima del límite superior de la normalidad o elevaciones de dos o más metabolitos.
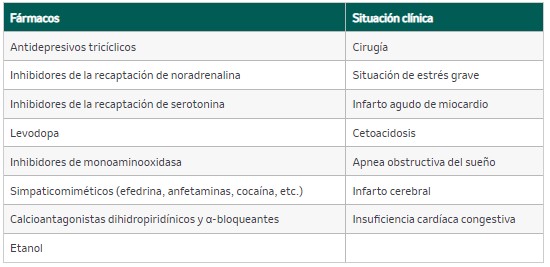
Tabla 19. Fármacos/situaciones que pueden incrementar los niveles de catecolaminas y metanefrinas (causas de falsos positivos)
5.3.2. Pruebas de imagen y diagnóstico de localización
5.3.2.1. Tomografía computarizada y resonancia magnética abdominopélvica
La tomografía computarizada (TC) con contraste se considera la prueba de primera elección por su mayor resolución espacial en tórax, abdomen y pelvis; la resonancia magnética (RM) se reserva para pacientes con alergia a contrastes yodados, niños y mujeres embarazadas.
5.3.2.2. Pruebas de Medicina Nuclear
Pueden ser útiles en pacientes con masas bilaterales o de gran tamaño, recurrencias o alto riesgo de enfermedad multifocal (síndromes hereditarios); la gammagrafía con 123I-metayodobencilguanidina (123I-MIBG) es la prueba de elección.
5.4. Tratamiento
El tratamiento de elección es quirúrgico. Previamente se debe realizar un manejo médico en todos los pacientes con los siguientes objetivos:
- Prevención de hipotensión posquirúrgica mediante la repleción de volumen; se recomienda una dieta rica en sal e ingesta hídrica abundante desde el segundo o tercer día de inicio del α-bloqueo.
- Prevención de crisis adrenérgica a través del bloqueo α-adrenérgico (fenoxibenzamina y doxazosina); se debe iniciar 7-14 días antes de la cirugía y siempre antes del bloqueo β-adrenérgico.
- Prevención de arritmias mediante el bloqueo β-adrenérgico (propranolol y atenolol), que controla la taquicardia refleja inducida tras el α-bloqueo; se debe iniciar 2-3 días antes de la cirugía, tras lograr un bloqueo α-adrenérgico adecuado.
En el postoperatorio pueden surgir complicaciones (principalmente arritmias, hipotensión arterial e hipoglucemia) que es necesario identificar precozmente y tratar de forma adecuada, por lo que se debe realizar una monitorización estrecha de la presión arterial, la frecuencia cardíaca y la glucemia en las primeras 24-48 h. Se ha de mantener una infusión intravenosa de suero salino fisiológico y suero glucosado durante las primeras 24 h.
6. Incidentaloma adrenal
6.1. Definición y etiología
El incidentaloma adrenal (IA) se define como cualquier masa adrenal asintomática de tamaño mayor o igual a 1 cm de diámetro detectada en pruebas de imagen no realizadas por sospecha de patología adrenal ni en el estudio de extensión de patología tumoral extraadrenal. En la tabla 20 se recogen las principales causas.
6.2. Diagnóstico
El estudio del IA tiene dos objetivos principales:
- Descartar malignidad mediante datos clínicos extraídos de la anamnesis y la exploración física y pruebas de imagen.
- Descartar funcionalidad mediante la historia clínica y los estudios bioquímico-hormonales (la secreción autónoma de cortisol es la forma de presentación más frecuente), si bien la mayoría son no funcionantes.
La tabla 21 incluye un resumen de las principales pruebas que se deben solicitar.
6.2.1. Pruebas de imagen
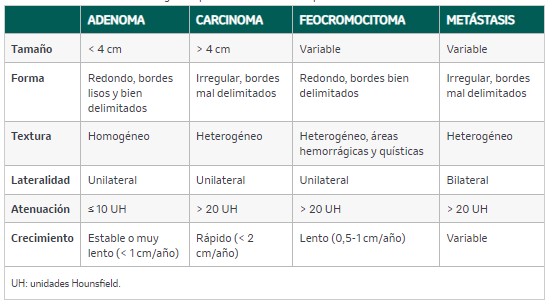
La TC sin contraste es la técnica de elección para la caracterización de los IA y permite diferenciar los adenomas (masas homogéneas con densidad menor de 10 unidades Hounsfield [UH]) de otro tipo de lesiones. En los casos en los que la lesión no puede caracterizarse mediante la TC sin contraste, pueden requerirse otras pruebas de imagen (TC con contraste, RM, tomografía por emisión de positrones [PET]-TC, gammagrafías, etc.). En la tabla 22 se recogen las características radiológicas de las lesiones suprarrenales más importantes.
Tabla 20. Etiología del incidentaloma adrenal
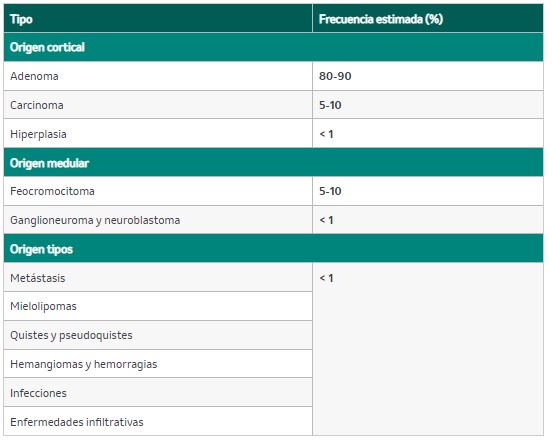
Tabla 21. Despistaje de funcionalidad en el incidentaloma adrenal
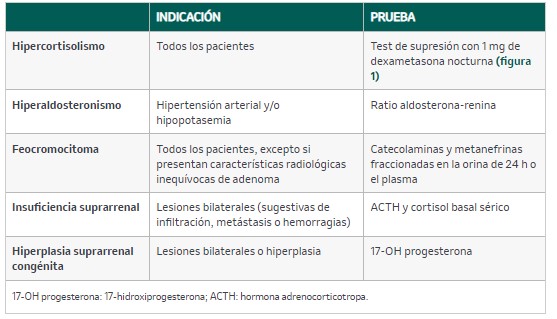
Tabla 22. Características radiológicas típicas de las lesiones suprarrenales
6.2.2. Punción-aspiración con aguja fina
Las indicaciones se limitan a pacientes con enfermedad oncológica conocida, cuando no se haya podido caracterizar la lesión por técnicas de imagen, y siempre que el resultado vaya a modificar la actitud terapéutica. Presenta rentabilidad limitada para el diagnóstico diferencial entre adenoma y carcinoma suprarrenal. Además, puede desencadenar una crisis adrenérgica si se trata de un feocromocitoma, por lo que siempre debe descartarse esta posibilidad.
6.3. Manejo
La cirugía es el tratamiento de elección en los IA funcionantes o con diagnóstico de malignidad y puede estar indicada en lesiones que no presentan características concluyentes de benignidad (masas unilaterales > 4 cm, hallazgos indeterminados o atípicos en pruebas de imagen, cambios en características radiológicas o crecimiento significativo).
Bibliografía
- Araujo-Castro M, Iturregui M, Calatayud M, Parra P, Gracia P, Alexandra F, et al. Practical guide on the initial evaluation, follow-up, and treatment of adrenal incidentalomas Adrenal Diseases Group of the Spanish Society of Endocrinology and Nutrition. Endocrinol Diabetes Nutr. 2020;67(6):408-419.
- Bornstein S, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and treatment of primary adrenal insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101(2):364-389.
- De Miguel P, Torres E, Palacios N, Moreira M, Solache I, Martínez ML, et al. Guía para el diagnóstico y tratamiento de la insuficiencia suprarrenal en el adulto. Endocrinol Nutr. 2014;6(Supl. 1)1:35.
- Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A, et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2016;175(2):G1-G34.
- Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101(5):1889-1916.
- Lenders JWM, Duh QY, Eisenhofer G, Giménez- Roqueplo A-P, Grebe SKG, Murad MH, et al. Phaeochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942.
- Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens. 2020;38(8):1443-1456.
- Santos S, Santos E, Gaztamide S, Salvador J. Diagnóstico y diagnóstico diferencial del síndrome de Cushing. Endocrinol Nutr. 2009;56(2):71-84.
- Sherlock M, Scarsbrook A, Abbas A, Fraser S, Limumpornpetch P, Dineen R, et al. Adrenal Incidentaloma. Endocr Rev. 2020;41(6):775-820.